Whippet.jl
Whippet


![]()
![]()

Graphical Overview
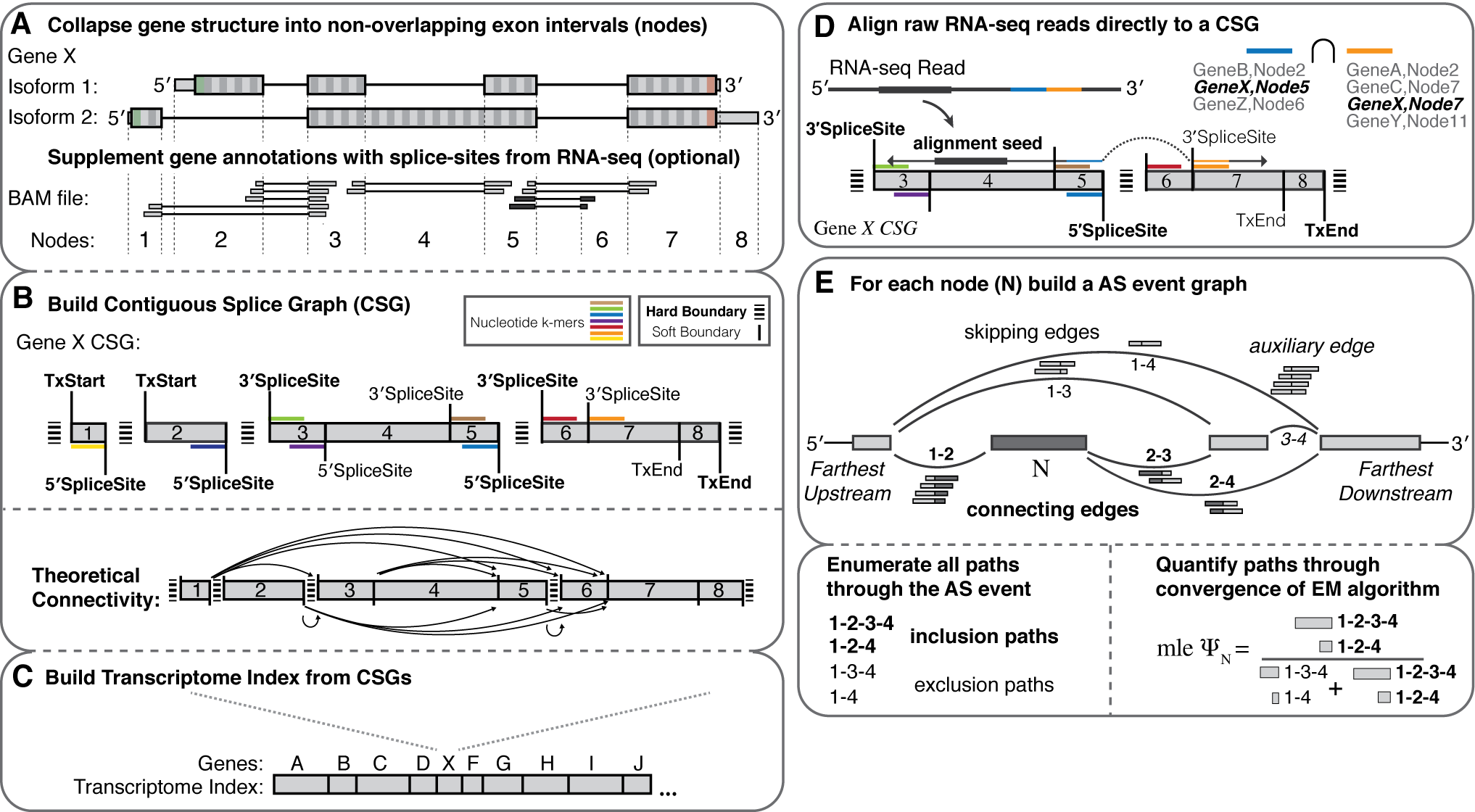
Features
- Splice graph representations of transcriptome structure
- Build an index for any species with a genome and annotation file
- Supplement the index with splice-sites/exons from independently aligned RNA-seq (BAM file).
- de novo AS event discovery (between indexed donor/acceptor splice sites)
- High speed PolyA+ Spliced Read Alignment (Read lengths <= 255)
- Repetitive read assignment for gene families
- Bias correction methods for 5’ sequence and GC-content
- On-the-fly alignment/re-analysis of SRR accession ids using ebi.ac.uk
- Fast and robust quantification of transcriptome structure and expression using EM
- Dynamic building and entropic measurements of splicing events of any complexity
- Percent-spliced-in (PSI) from event-level EM
- Gene expression (TPM) from transcript-level EM
- Differential splicing comparisons
- Probabilistic calculations of delta PSI leveraging multi-sample biological replicates
Paper: https://doi.org/10.1016/j.molcel.2018.08.018
Get started
1) Install
Whippet v1.6 works on the long-term support release of Julia (v1.6.7) which is available here (https://julialang.org). (Note: Whippet.jl does not yet work on Julia v1.9). If you are new to julia, there is a helpful guide on how to get it up and running here
Download and install dependencies
git clone https://github.com/timbitz/Whippet.jl.git
cd Whippet.jl
julia --project -e 'using Pkg; Pkg.instantiate(); Pkg.test()'
This should tell you Testing Whippet tests passed
Notes:
- Everything in Whippet.jl/bin should work out-of-the-box, however the first time running will be slow as julia will be precompiling code
- Update to the most recent version of Whippet by pulling the master branch
git pull - For all executables in
Whippet.jl/bin, you can use the-hflag to get a list of the available command line options, their usage and defaults. - You should install Julia locally, if you have to install system-wide, there is some help here
- For instructions on using Whippet with Julia v0.6.4, look at the README.md within the Whippet v0.11.1 tag (but please note this verison is no longer supported)
2) Build an index.
a) Annotation (GTF) only index.
You need your genome sequence in fasta, and a gene annotation file in Ensembl-style GTF format. Default GENCODE annotation supplied for hg19 and mm10 in Whippet/anno. You can also obtain Ensembl GTF files from these direct links for Human: Ensembl_hg38_release_92 and Mouse: Ensembl_mm10_release_92. Other Ensembl GTF files can be downloaded here.
Download the genome.
wget https://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/hg19.fa.gz
Build an index.
julia bin/whippet-index.jl --fasta hg19.fa.gz --gtf anno/gencode_hg19.v25.tsl1.gtf.gz
Notes:
- Whippet only uses GTF
exonlines (others are ignored). These must contain bothgene_idandtranscript_idattributes (which should not be the same as one another!). This GTF file should be consistent with the GTF2.2 specification, and should have all entries for a transcript in a continuous block. Warning: The UCSC table browser will not produce valid GTF2.2 files. Similarly, GTF files obtained from iGenomes or the Refseq websites do not satisfy these specifications. - You can specify the output name and location of the index to build using the
-x / --indexparameter. The default (for both whippet-index.jl and whippet-quant.jl) is a generic index namedgraphlocated atWhippet.jl/index/graph.jls, so you must have write-access to this location to use the default.
b) Annotation (GTF) + Alignment (BAM) supplemented index.
Whippet v0.11+ allows you to build an index that includes unannotated splice-sites and exons found in a spliced RNA-seq alignment file. In order to build a BAM supplemented index, you need your BAM file sorted and indexed (using samtools):
# If using multiple BAM files (tissue1, ..., tissue3 etc), merge them first:
samtools merge filename.bam tissue1.bam tissue2.bam tissue3.bam
# If using a single BAM file start here:
samtools sort -o filename.sort filename.bam
samtools rmdup -S filename.sort.bam filename.sort.rmdup.bam
samtools index filename.sort.rmdup.bam
ls filename.sort.rmdup.bam*
filename.sort.rmdup.bam filename.sort.rmdup.bam.bai
Then build an index but with the additional --bam parameter:
julia bin/whippet-index.jl --fasta hg19.fa.gz --bam filename.sort.rmdup.bam --gtf anno/gencode_hg19.v25.tsl1.gtf.gz
Notes:
- The
--bamoption is sensitive to alignment strand, therefore using strand-specific alignments is recommended. - By default only spliced alignments where one of the splice-sites match a known splice-site in the annotation are used, to reduce false positives due to overlapping gene regions (i.e. falsely adding splice sites that belong to a different, but overlapping gene, which is common in many species). Use the
--bam-both-novelflag to override this requirement for greater Recall of unannotated splice-sites. - Control the minimum number of reads required to consider a novel splice site from BAM using the
--bam-min-readsparameter (default is 1). Increase this parameter with large bam files to reduce artifacts and one-off cryptic splice junctions.
3) Quantify FASTQ files.
a) Single-end reads
julia bin/whippet-quant.jl file.fastq.gz
Note: Whippet only accepts standard four-line FASTQ file (described here: https://support.illumina.com/bulletins/2016/04/fastq-files-explained.html)
Also, as of version 1.0.0, --ebi and --url flags have been deprecated to ease maintenance. EBI file paths can be found at the URL http://www.ebi.ac.uk/ena/data/warehouse/filereport?accession=$ebi_id&result=read_run&fields=fastq_ftp&display=txt. Use your own accession id (SRR id) in place of $ebi_id.
For example:
curl "https://www.ebi.ac.uk/ena/data/warehouse/filereport?accession=SRR1199010&result=read_run&fields=fastq_ftp&display=txt"
fastq_ftp
ftp.sra.ebi.ac.uk/vol1/fastq/SRR119/000/SRR1199010/SRR1199010.fastq.gz
b) Paired-end reads
julia bin/whippet-quant.jl fwd_file.fastq.gz rev_file.fastq.gz
To locate paired-end SRR id files, use the same ebi.ac.uk URL:
curl "https://www.ebi.ac.uk/ena/data/warehouse/filereport?accession=ERR1994736&result=read_run&fields=fastq_ftp&display=txt"
fastq_ftp
ftp.sra.ebi.ac.uk/vol1/fastq/ERR199/006/ERR1994736/ERR1994736_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/ERR199/006/ERR1994736/ERR1994736_2.fastq.gz
c) Non-default input/output
To specify output location or a specific index:
julia bin/whippet-quant.jl fwd_file.fastq.gz -o outputname -x customindex.jls
You can also output the alignments in SAM format with the --sam flag and convert to bam with samtools:
julia bin/whippet-quant.jl fwd_file.fastq.gz --sam > fwd_file.sam
samtools view -bS fwd_file.sam > fwd_file.bam
For greater stability of quantifications across multiple RNA-seq protocols, try the --biascorrect flag, which will apply GC-content and 5’ sequence bias correction methods:
julia bin/whippet-quant.jl fwd_file.fastq.gz --biascorrect
It is also possible to pool fastq files at runtime using shell commands, and the optional (--force-gz) for pooled gz files (files without .gz suffix)
julia bin/whippet-quant.jl <( cat time-series_{1,2,3,4,5}.fastq.gz ) --force-gz -o interval_1-5
4) Compare multiple psi files
Compare .psi.gz files from from two samples -a and -b with any number of replicates (comma delimited list of files or common pattern matching) per sample.
ls *.psi.gz
#sample1-r1.psi.gz sample1-r2.psi.gz sample2-r1.psi.gz sample2-r2.psi.gz
julia bin/whippet-delta.jl -a sample1 -b sample2
#OR
julia bin/whippet-delta.jl -a sample1-r1.psi.gz,sample1-r2.psi.gz -b sample2-r1.psi.gz,sample2-r2.psi.gz
Note: comparisons of single files still need a comma: -a singlefile_a.psi.gz, -b singlefile_b.psi.gz,
Output Formats
The output format for whippet-quant.jl is saved into two core quant filetypes, .psi.gz and .tpm.gz files.
Each .tpm.gz file contains a simple format compatible with many downstream tools (one for the TPM of each annotated transcript, and another at the gene-level):
| Gene/Isoform | TpM | Read Counts |
|---|---|---|
| NFIA | 2897.11 | 24657.0 |
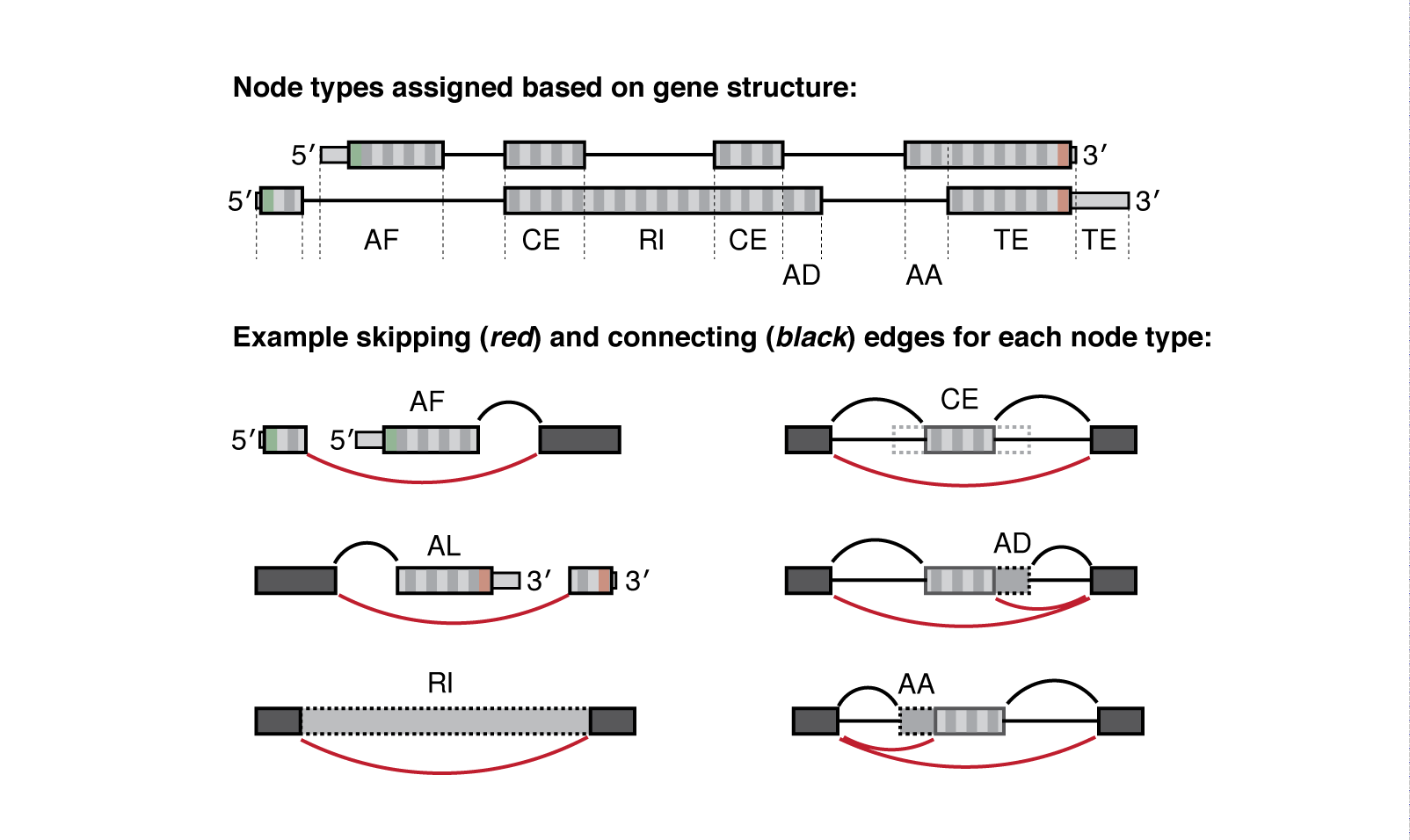
Meanwhile the .psi.gz file is a bit more complex and requires more explanation. Quantified nodes can fall into a number of “node-type” categories:
| Type | Interpretation |
|---|---|
| CE | Core exon, which may be bounded by one or more alternative AA/AD nodes |
| AA | Alternative Acceptor splice site |
| AD | Alternative Donor splice site |
| RI | Retained intron |
| TS | Tandem transcription start site |
| TE | Tandem alternative polyadenylation site |
| AF | Alternative First exon |
| AL | Alternative Last exon |
| BS | Circular back-splicing (output only when --circ flag is given) |
Each node is defined by a type (above) and has a corresponding value for Psi or the Percent-Spliced-In, see below:
The .psi.gz file itself is outputted in the form:
| Gene | Node | Coord | Strand | Type | Psi | CI Width | CI Lo,Hi | Total Reads | Complexity | Entropy | Inc Paths | Exc Paths |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NFIA | 2 | chr1:61547534-61547719 | + | AF | 0.782 | 0.191 | 0.669,0.86 | 49.0 | K1 | 0.756 | 2-4-5:0.782 | 1-5:0.218 |
| NFIA | 4 | chr1:61548433-61548490 | + | CE | 0.8329 | 0.069 | 0.795,0.864 | 318.0 | K2 | 1.25 | 2-4-5:0.3342,3-4-5:0.4987 | 1-5:0.1671 |
| NFIA | 5 | chr1:61553821-61554352 | + | CE | 0.99 | NA | NA | NA | NA | NA | NA | NA |
| NFIA | 6 | chr1:61743192-61743257 | + | CE | 0.99 | NA | NA | NA | NA | NA | NA | NA |
Whippet outputs quantification at the node-level (one PSI value for each node, not exon). For example, Whippet CE (‘core exon’) nodes may be bounded by one or more AA or AD nodes. So when those AA or AD nodes are spliced in, the full exon consists of the nodes combined (AA+CE).
We believe that this is (and should be) the correct generalization of event-level splicing quantification for the following reasons:
- Whippet allows for dynamic quantification of observed splicing patterns. Therefore, in order to maintain consistent output from different Whippet runs on various samples, the basic unit of quantification needs to be a
node. - Whippet can handle highly complex events, in contrast to many other splicing quantification tools which only report binary event types. Since, for example, it is possible for both (CE+AA) and CE exons to be excluded from the mature message, complex events may involve a number of overlapping full exons. If Whippet output was enumerated for all such combinations, the output for complex events would grow exponentially and approach uselessness.
- The general definition of Percent-spliced-in for an exon (or node) is the percentage of transcripts that ‘have that exon spliced in’, irrespective of the upstream or downstream splice sites that connect to it (those merely alter another node’s PSI). Therefore, we feel that the output of PSI values should not change based on upstream or downstream splice-sites as they might with some other programs.
This must be taken into account when intersecting .psi.gz coordinate output with other formats that only represent full exons (which can be one or more adjacent nodes combined). To ease this process, whippet-index.jl outputs an index.exons.gz file that contains all theoretical full exon coordinates mapped to Whippet nodes for each gene, and whether or not the exon is found in annotation.
Note:
- Complexity refers to the discrete categories based-on the ceiling(log2(number of paths)) through each AS event.
- Entropy refers to the shannon-entropy of the relative expression of the paths through the AS event.
- Inc_Paths/Exc_Paths contain the paths quantified through the AS event (supporting inclusion or exclusion respectively), each path (eg. 1-2-3) gives the set of nodes in the path, followed by a
:and the relative expression, such that the sum of all these paths is 1.0.
The raw junctions are output in the format of .jnc.gz files, which look like:
| Chrom | Donor_Coord | Acceptor_Coord | GeneID:Nodes:Type | ReadCount | Strand |
|---|---|---|---|---|---|
| chr1 | 150600068 | 150601890 | ENSG00000143420.17_1:2-3:CON_ANNO | 113.04 | - |
| chr1 | 150598284 | 150599943 | ENSG00000143420.17_1:3-6:ALT_ANNO | 110.12 | - |
| chr1 | 150595335 | 150598118 | ENSG00000143420.17_1:6-9:CON_ANNO | 35.74 | - |
| chr1 | 161185160 | 161187776 | ENSG00000158869.10_1:1-2:CON_ANNO | 20.47 | + |
| chr1 | 161185160 | 161188031 | ENSG00000158869.10_1:1-3:ALT_UNIQ | 3.11 | + |
The Type can be one of three:
| Type | Meaning |
|---|---|
| CON_ANNO | Annotated constitutive junction |
| ALT_ANNO | Annotated alternative junction |
| ALT_UNIQ | Unannotated alternative junction |
The output format of .diff.gz files from whippet-delta.jl is:
| Gene | Node | Coord | Strand | Type | Psi_A | Psi_B | DeltaPsi | Probability | Complexity | Entropy |
|---|---|---|---|---|---|---|---|---|---|---|
| ENSG00000117448.13_2 | 9 | chr1:46033654-46033849 | + | CE | 0.95971 | 0.97876 | -0.019047 | 0.643 | K0 | 0.0 |
| ENSG00000117448.13_2 | 10 | chr1:46034157-46034356 | + | CE | 0.9115 | 0.69021 | 0.22129 | 0.966 | K1 | 0.874 |
Between the set of replicates from -a and -b whippet-delta.jl outputs a mean Psi_A (from emperical sampling of the joint posterior distribution) and mean Psi_B. It also outputs the mean deltaPsi (Psi_A - Psi_B) and the probability that there is some change in deltaPsi given the read depth of the AS event, such that P( |
deltaPsi | > 0.0). To determine the significance of output in .diff.gz files, two heuristic filters are encouraged, however please note that the exact values for these filters have not yet been systematically optimized. These should be adjusted by the user according to the desired stringency, read depth, and experiment set-up: (1) In general, significant nodes should have a higher probability (e.g. >=0.90) suggesting there is likely to be a difference between the samples, and (2) significant nodes should have an absolute magnitude of change ( |
DeltaPsi | > x) where x is some value you consider to be a biologically relevant magnitude of change (we suggest | DeltaPsi | > 0.1). |
Visualization
Currently, Whippet supports the output of RNA-seq alignments in SAM/BAM format when the (--sam) flag is used. These alignment files can be displayed using a number of different downstream visualization software tools, such as the IGV browser, which also supports display in the popular “sashimi-plot” format (e.g. instructions on making sashimi-plots with IGV can be found here).
Index Building Strategies
If you are building an index for another organism, there are some general guidelines that can help to ensure that the index you build is as effective as it can be. In general you should seek to:
- Use only the highest quality annotations you can find.
- Use strand-specific RNA-seq alignments to avoid false positives from overlapping gene regions (eg. head-to-head and tail-to-tail)
- If a high-quality annotation set like GENCODE, contains both high and low quality transcripts, filter it! For human, we only use TSL-1/2 annotations.
- Avoid giving annotations with ‘indels’ such as ESTs or mRNAs without filtering out invalid splice sites first.
- If you plan to align very short reads (~36nt), consider decreasing the Kmer size (we have used 6nt before, though this is quite low and it is not recommended to go below this), otherwise the default Kmer size (9nt) should be used. However, reads of lower length should still be of high-quality to expect performant spliced-read alignment.
Troubleshooting
With all of the executables in Whippet/bin, you can use the -h flag to get a list of the available command line options and their usage. If you are having trouble using or interpreting the output of Whippet then please ask a question in our gitter chat!.
If you are having trouble running a Whippet executable in /bin, try opening the julia REPL within the Whippet.jl directory and testing:
]
(@v1.5) pkg> activate .
(Whippet) pkg> test
Whippet should run cleanly if your environment is working correctly (although it is mostly untested on Windows, it should still work– or at least it did the one time we tried it ;-). If you still think you have found a bug feel free to open an issue in github or make a pull request!